

Fusion, the term generally applied to the melting of a solid substance, or the change of state of aggregation from the solid to the liquid. The term “liquefaction” is frequently employed in the same sense, but is often restricted to the condensation of a gas or vapour. The converse process of freezing or solidification, the change from the liquid to the solid state, is subject to the same laws, and must be considered together with fusion. The solution of a solid in a foreign liquid, and the deposition or crystallization of a solid from a solution, are so closely related to the fusion of a pure substance, that it will also be necessary to consider some of the analogies which they present.
1. General Phenomena.—There are two chief varieties of the process of fusion, namely, crystalline and amorphous, which are in many ways distinct, although it is possible to find intermediate cases which partake of the characteristics of both. The melting of ice may be taken as a typical case of crystalline fusion. The passage from rigid solid to mobile liquid occurs at a definite surface without any intermediate stage or plastic condition. The change takes place at a definite temperature, the fusing or freezing point (abbreviated F.P.), and requires the addition of a definite quantity of heat to the solid, which is called the latent heat of fusion. There is also in general a considerable change of volume during fusion, which amounts in the case of ice to a contraction of 9%. Typical cases of amorphous solidification are those of silica, glass, plastic sulphur, pitch, alcohol and many organic liquids. In this type the liquid gradually becomes more and more viscous as the temperature falls, and ultimately attains the rigidity characteristic of a solid, without any definite freezing point or latent heat. The condition of the substance remains uniform throughout, if its temperature is uniform; there is no separation into the two distinct phases of solid and liquid, and there is no sudden change of volume at any temperature.
A change or transition from one crystalline form to another may occur in the solid state with evolution or absorption of heat at a definite temperature, and is analogous to the change from solid to liquid, but usually takes place more slowly owing to the small molecular mobility of the solid state. Thus rhombic sulphur when heated passes slowly at 95.6° C. into the monosymmetric form which melts at 120°, but if heated rapidly the rhombic form melts at 114.5. The two forms, rhombic and monosymmetric, can exist in equilibrium at 95.6°, the transition point at which they have the same vapour pressure. Similarly a solid solution of carbon in iron, when cooled slowly, passes at about 700° C., with considerable evolution of heat, into the form of “pearlite,” which is soft when cold, but if rapidly chilled the carbon remains in solution and the steel is very hard (see also Alloys).
In the case of crystalline fusion it is necessary to distinguish two cases, the homogeneous and the heterogeneous. In the first case the composition of the solid and liquid phases are the same, and the temperature remains constant during the whole process of fusion. In the second case the solid and liquid phases differ in composition; that of the liquid phase changes continuously, and the temperature does not remain constant during the fusion. The first case comprises the fusion of pure substances, and that of eutectics, or cryohydrates; the second is the general case of an alloy or a solution. These have been very fully studied and their phenomena greatly elucidated in recent years.
There is also a sub-variety of amorphous fusion, which may be styled colloid or gelatinous, and may be illustrated by the behaviour of solutions of water in gelatin. Many of these jellies melt at a fairly definite temperature on heating, and coagulate or set at a definite temperature on cooling. But in some cases the process is not reversible, and there is generally marked hysteresis, the temperature of setting and other phenomena depending on the rate of cooling. This case has not yet been fully worked out; but it appears probable that in many cases the jelly possesses a spongy framework of solid, holding liquid in its meshes or interstices. It might be regarded as a case of “heterogeneous” amorphous fusion, in which the liquid separates into two phases of different composition, one of which solidifies before the other. The two phases cannot, as a rule, be distinguished optically, but it is generally possible to squeeze out some of the liquid phase when the jelly has set, which proves that the substance is not really homogeneous. In very complicated mixtures, such as acid lavas or slags containing a large proportion of silica, amorphous and crystalline solidification may occur together. In this case the crystals separate first during the process of cooling, the mother liquor increases gradually in viscosity, and finally sets as an amorphous ground-mass or matrix, in which crystals of different kinds and sizes, formed at different stages of the cooling, remain embedded. The formation of crystals in an amorphous solid after it has set is also of frequent occurrence. It is termed devitrification, but is a very slow process unless the solid is in a plastic state.
2. Homogeneous Crystalline Fusion.—The fusion of a solid of this type is characterized most clearly by the perfect constancy of temperature during the process. In fact, the law of constant temperature, which is generally stated as the first of the so-called “laws of fusion,” does not strictly apply except to this case. The constancy of the F.P. of a pure substance is so characteristic that change of the F.P. is often one of the most convenient tests of the presence of foreign material. In the case of substances like ice, which melt at a low temperature and are easily obtained in large quantities in a state of purity, the point of fusion may be very accurately determined by observing the temperature of an intimate mixture of the solid and liquid while slowly melting as it absorbs heat from surrounding bodies. But in the majority of cases it is more convenient to observe the freezing point as the liquid is cooled. By this method it is possible to ensure perfect uniformity of temperature throughout the mass by stirring the liquid continuously during the process of freezing, whereas it is difficult to ensure uniformity of temperature in melting a solid, however gradually the heat is supplied, unless the solid can be mixed with the liquid. It is also possible to observe the F.P. in other ways, as by noting the temperature at the moment of the breaking of a wire, of the stoppage of a stirrer, or of the maximum rate of change of volume, but these methods are generally less certain in their indications than the point of greatest constancy of temperature in the case of homogeneous crystalline solids.
Fusing Points of Common Metals
| Mercury | −38.8° | Antimony | 630° |
| Potassium | 62.5° | Aluminium | 655° |
| Sodium | 95.6° | Silver | 962° |
| Tin | 231.9° | Gold | 1064° |
| Bismuth | 269.2° | Copper | 1082° |
| Cadmium | 320.7° | Nickel | 1427° |
| Lead | 327.7° | Palladium | 1535° |
| Zinc | 419.0° | Platinum | 1710° |
The above table contains some of the most recent values of fusing points of metals determined (except the first three and the last three) with platinum thermometers. The last three values are those obtained by extrapolation with platinum-rhodium and platinum-iridium couples. (See Harker, Proc. Roy. Soc. A 76, p. 235, 1905.) Some doubt has recently been raised with regard to the value for platinum, which is much lower than that previously accepted, namely 1775°.
3. Superfusion, Supersaturation.—It is generally possible to cool a liquid several degrees below its normal freezing point without a separation of crystals, especially if it is protected from agitation, which would assist the molecules to rearrange themselves. A liquid in this state is said to be “undercooled” or “superfused.” The phenomenon is even more familiar in the case of solutions (e.g. sodium sulphate or acetate) which may remain in the “metastable” condition for an indefinite time if protected from dust, &c. The introduction into the liquid under this condition of the smallest fragment of the crystal, with respect to which the solution is supersaturated, will produce immediate crystallization, which will continue until the temperature is raised to the saturation point by the liberation of the latent heat of fusion. The constancy of temperature at the normal freezing point is due to the equilibrium of exchange existing between the liquid and solid. Unless both solid and liquid are present, there is no condition of equilibrium, and the temperature is indeterminate.
It has been shown by H.A. Miers (Jour. Chem. Soc., 1906, 89, p. 413) that for a supersaturated solution in metastable equilibrium there is an inferior limit of temperature, at which it passes into the “labile” state, i.e. spontaneous crystallization occurs throughout the mass in a fine shower. This seems to be analogous to the fine misty condensation which occurs in a supersaturated vapour in the absence of nuclei (see Vaporization) when the supersaturation exceeds a certain limit.
4. Effect of Pressure on the F.P.—The effect of pressure on the fusing-point depends on the change of volume during fusion. Substances which expand on freezing, like ice, have their freezing points lowered by increase of pressure; substances which expand on fusing, like wax, have their melting points raised by pressure. In each case the effect of pressure is to retard increase of volume. This effect was first predicted by James Thomson on the analogy of the effect of pressure on the boiling point, and was numerically verified by Lord Kelvin in the case of ice, and later by Bunsen in the case of paraffin and spermaceti. The equation by which the change of the F.P. is calculated may be proved by a simple application of the Carnot cycle, exactly as in the case of vapour and liquid. (See Thermodynamics.) If L be the latent heat of fusion in mechanical units, v′ the volume of unit mass of the solid, and v″ that of the liquid, the work done in an elementary Carnot cycle of range dθ will be dp(v″ − v′), if dp is the increase of pressure required to produce a change dθ in the F.P. Since the ratio of the work-difference or cycle-area to the heat-transferred L must be equal to dθ/θ, we have the relation
dθ/dp = θ (v″ − v′)/L.
The sign of dθ, the change of the F.P., is the same as that of the change of volume (v″ − v′). Since the change of volume seldom exceeds 0.1 c.c. per gramme, the change of the F.P. per atmosphere is so small that it is not as a rule necessary to take account of variations of atmospheric pressure in observing a freezing point. A variation of 1 cm. in the height of the barometer would correspond to a change of .0001° C. only in the F.P. of ice. This is far beyond the limits of accuracy of most observations. Although the effect of pressure is so small, it produces, as is well known, remarkable results in the motion of glaciers, the moulding and regelation of ice, and many other phenomena. It has also been employed to explain the apparent inversion of the order of crystallization in rocks like granite, in which the arrangement of the crystals indicates that the quartz matrix solidified subsequently to the crystals of felspar, mica or hornblende embedded in it, although the quartz has a higher melting point. It is contended that under enormous pressure the freezing points of the more fusible constituents might be raised above that of the quartz, if the latter is less affected by pressure. Thus Bunsen found the F.P. of paraffin wax 1.4° C. below that of spermaceti at atmospheric pressure. At 100 atmospheres the two melted at the same temperature. At higher pressures the paraffin would solidify first. The effect of pressure on the silicates, however, is much smaller, and it is not so easy to explain a change of several hundred degrees in the F.P. It seems more likely in this particular case that the order of crystallization depends on the action of superheated water or steam at high temperatures and pressures, which is well known to exert a highly solvent and metamorphic action on silicates.
5. Variation of Latent Heat.—C.C. Person in 1847 endeavoured to show by the application of the first law of thermodynamics that the increase of the latent heat per degree should be equal to the difference (s″ − s′) between the specific heats of the liquid and solid. If, for instance, water at 0° C. were first frozen and then cooled to −t° C., the heat abstracted per gramme would be (L′ + s′t) calories. But if the water were first cooled to −t° C., and then frozen at −t°C., by abstracting heat L″, the heat abstracted would be L″ + s″t. Assuming that the heat abstracted should be the same in the two cases, we evidently obtain L′ − L″ = (s″ − s′)t. This theory has been approximately verified by Petterson, by observing the freezing of a liquid cooled below its normal F.P. (Jour. Chem. Soc. 24, p. 151). But his method does not represent the true variation of the latent heat with temperature, since the freezing, in the case of a superfused liquid, really takes place at the normal freezing point. A quantity of heat s″t is abstracted in cooling to −t, (L″ − s″t) in raising to 0° and freezing at 0°, and s’t in cooling the ice to -t. The latent heat L″ at −t does not really enter into the experiment. In order to make the liquid freeze at a different temperature, it is necessary to subject it to pressure, and the effect of the pressure on the latent heat cannot be neglected. The entropy of a liquid φ″ at its F.P. reckoned from any convenient zero φ0 in the solid state may be represented by the expression
φ″ − φ0 = ∫ s′dθ/θ + L/θ.
Since θdφ″/dθ = s″, we obtain by differentiation the relation
dL/dθ = s″ − s′ + L/θ,
which is exactly similar to the equation for the specific heat of a vapour maintained in the saturated condition. If we suppose that the specific heats s′ and s″ of the solid and liquid at equilibrium pressure are nearly the same as those ordinarily observed at constant pressure, the relation (3) differs from that of Person only by the addition of the term L/θ. Since s″ is greater than s′ in all cases hitherto investigated, and L/θ is necessarily positive, it is clear that the latent heat of fusion must increase with rise of temperature, or diminish with fall of temperature. It is possible to imagine the F.P. so lowered by pressure (positive or negative) that the latent heat should vanish, in which case we should probably obtain a continuous passage from the liquid to the solid state similar to that which occurs in the case of amorphous substances. According to equation (3), the rate of change of the latent heat of water is approximately 0.80 calorie per degree at 0° C. (as compared with 0.50, Person), if we assume s″ = 1, and s′ = 0.5. Putting (s″ − s′) = 0.5 in equation (2), we find L = 0 at −160° C. approximately, but no stress can be laid on this estimate, as the variation of (s″ − s′) is so uncertain.
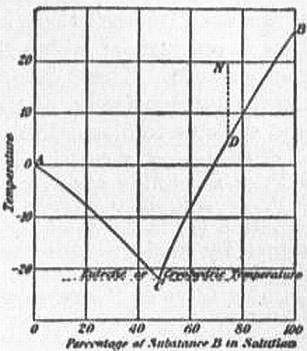 |
| Fig. 1.—F.P. or Solubility Curve: simple case. |
6. Freezing of Solutions and Alloys.—The phenomena of freezing of heterogeneous crystalline mixtures may be illustrated by the case of aqueous solutions and of metallic solutions or alloys, which have been most widely studied. The usual effect of an impurity, such as salt or sugar in solution in water, is to lower the freezing point, so that no crystallization occurs until the temperature has fallen below the normal F.P. of the pure solvent, the depression of F.P. being nearly proportional to the concentration of the solution. When freezing begins, the solvent generally separates out from the solution in the pure state. This separation of the solvent involves an increase in the strength of the remaining solution, so that the temperature does not remain constant during the freezing, but continues to fall as more of the solvent is separated. There is a perfectly definite relation between temperature and concentration at each stage of the process, which may be represented in the form of a curve as AC in fig. 1, called the freezing point curve. The equilibrium temperature, at the surface of contact between the solid and liquid, depends only on the composition of the liquid phase and not at all on the quantity of solid present. The abscissa of the F.P. curve represents the composition of that portion of the original solution which remains liquid at any temperature. If instead of starting with a dilute solution we start with a strong solution represented by a point N, and cool it as shown by the vertical line ND, a point D is generally reached at which the solution becomes “saturated.” The dissolved substance or “solute” then separates out as the solution is further cooled, and the concentration diminishes with fall of temperature in a definite relation, as indicated by the curve CB, which is called the solubility curve. Though often called by different names, the two curves AC and CB are essentially of a similar nature. To take the case of an aqueous solution of salt as an example, along CB the solution is saturated with respect to salt, along AC the solution is saturated with respect to ice. When the point C is reached along either curve, the solution is saturated with respect to both salt and ice. The concentration cannot vary further, and the temperature remains constant, while the salt and ice crystallize out together, maintaining the exact proportions in which they exist in the solution. The resulting solid was termed a cryohydrate by F. Guthrie, but it is really an intimate mixture of two kinds of crystals, and not a chemical compound or hydrate containing the constituents in chemically equivalent proportions. The lowest temperature attainable by means of a freezing mixture is the temperature of the F.P. of the corresponding cryohydrate. In a mixture of salt and ice with the least trace of water a saturated brine is quickly formed, which dissolves the ice and falls rapidly in temperature, owing to the absorption of the latent heat of fusion. So long as both ice and salt are present, if the mixture is well stirred, the solution must necessarily become saturated with respect to both ice and salt, and this can only occur at the cryohydric temperature, at which the two curves of solubility intersect.
The curves in fig. 1 also illustrate the simplest type of freezing point curve in the case of alloys of two metals A and B which do not form mixed crystals or chemical compounds. The alloy corresponding to the cryohydrate, possessing the lowest melting point, is called the eutectic alloy, as it is most easily cast and worked. It generally possesses a very fine-grained structure, and is not a chemical compound. (See Alloys.)
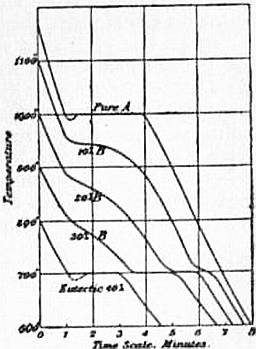 |
| Fig. 2.—Cooling Curves of Alloys: typical case. |
To obtain a complete F.P. curve even for a binary alloy is a laborious and complicated process, but the information contained in such a curve is often very valuable. It is necessary to operate with a number of different alloys of suitably chosen composition, and to observe the freezing points of each separately. Each alloy should also be analysed after the process if there is any risk of its composition having been altered by oxidation or otherwise. The freezing points are generally best determined by observing the gradual cooling of a considerable mass, which is well stirred so long as it remains liquid. The curve of cooling may most conveniently be recorded, either photographically, using a thermocouple and galvanometer, as in the method of Sir W. Roberts-Austen, or with pen and ink, if a platinum thermometer is available, according to the method put in practice by C.T. Heycock and F.H. Neville. A typical set of curves obtained in this manner is shown in fig. 2. When the pure metal A in cooling reaches its F.P. the temperature suddenly becomes stationary, and remains accurately constant for a considerable period. Often it falls slightly below the F.P. owing to super-fusion, but rises to the F.P. and remains constant as soon as freezing begins. The second curve shows the cooling of A with 10% of another metal B added. The freezing begins at a lower temperature with the separation of pure A. The temperature no longer remains constant during freezing, but falls more and more rapidly as the proportion of B in the liquid increases. When the eutectic temperature is reached there is a second F.P. or arrest at which the whole of the remaining liquid solidifies. With 20% of B the first F.P. is further lowered, and the temperature falls faster. The eutectic F.P. is of longer duration, but still at the same temperature. For an alloy of the composition of the eutectic itself there is no arrest until the eutectic temperature is reached, at which the whole solidifies without change of temperature. There is a great advantage in recording these curves automatically, as the primary arrest is often very slight, and difficult to observe in any other way.
7. Change of Solubility with Temperature.—The lowering of the F.P. of a solution with increase of concentration, as shown by the F.P. or solubility curves, may be explained and calculated by equation (1) in terms of the osmotic pressure of the dissolved substance by analogy with the effect of mechanical pressure. It is possible in salt solutions to strain out the salt mechanically by a suitable filter or “semi-permeable membrane,” which permits the water to pass, but retains the salt. To separate 1 gramme of salt requires the performance of work PV against the osmotic pressure P, where V is the corresponding diminution in the volume of the solution. In dilute solutions, to which alone the following calculation can be applied, the volume V is the reciprocal of the concentration C of the solution in grammes per unit volume, and the osmotic pressure P is equal to that of an equal number of molecules of gas in the same space, and may be deduced from the usual equation of a gas,
P = Rθ / VM = RθC / M,
where M is the molecular weight of the salt in solution, θ the absolute temperature, and R a constant which has the value 8.32 joules, or nearly 2 calories, per degree C. It is necessary to consider two cases, corresponding to the curves CB and AB in fig. 1, in which the solution is saturated with respect to salt and water respectively. To facilitate description we take the case of a salt dissolved in water, but similar results apply to solutions in other liquids and alloys of metals.
(a) If unit mass of salt is separated in the solid state from a saturated solution of salt (curve CB) by forcing out through a semi-permeable membrane against the osmotic pressure P the corresponding volume of water V in which it is dissolved, the heat evolved is the latent heat of saturated solution of the salt Q together with the work done PV. Writing (Q + PV) for L, and V for (v″ − v′) in equation (1), and substituting P for p, we obtain
Q + PV = VθdP / dθ,
which is equivalent to equation (1), and may be established by similar reasoning. Substituting for P and V in terms of C from equation (4), if Q is measured in calories, R = 2, and we obtain
QC = 2θ²dC / dθ,
which may be integrated, assuming Q constant, with the result
2logeC″ / C′ = Q / θ′ − Q / θ″,
where C′, C″ are the concentrations of the saturated solution corresponding to the temperatures θ′ and θ″. This equation may be employed to calculate the latent heat of solution Q from two observations of the solubility. It follows from these equations that Q is of the same sign as dC/dθ, that is to say, the solubility increases with rise of temperature if heat is absorbed in the formation of the saturated solution, which is the usual case. If, on the other hand, heat is liberated on solution, as in the case of caustic potash or sulphate of calcium, the solubility diminishes with rise of temperature.
(b) In the case of a solution saturated with respect to ice (curve AC), if one gramme of water having a volume v is separated by freezing, we obtain a precisely similar equation to (5), but with L the latent heat of fusion of water instead of Q, and v instead of V. If the solution is dilute, we may neglect the external work Pv in comparison with L, and also the heat of dilution, and may write P/t for dP/dθ, where t is the depression of the F.P. below that of the pure solvent. Substituting for P in terms of V from equation (4), we obtain
t = 2θ²v / LVM = 2θ²w / LWM,
where W is the weight of water and w that of salt in a given volume of solution. If M grammes of salt are dissolved in 100 of water, w = M and W = 100. The depression of the F.P. in this case is called by van ‘t Hoff the “Molecular Depression of the F.P.” and is given by the simple formula
t = .02θ² / L.
Equation (8) may be used to calculate L or M, if either is known, from observations of t, θ and w/W. The results obtained are sufficiently approximate to be of use in many cases in spite of the rather liberal assumptions and approximations effected in the course of the reasoning. In any case the equations give a simple theoretical basis with which to compare experimental data in order to estimate the order of error involved in the assumptions. We may thus estimate the variation of the osmotic pressure from the value given by the gaseous equation, as the concentration of the solution or the molecular dissociation changes. The most uncertain factor in the formula is the molecular weight M, since the molecule in solution may be quite different from that denoted by the chemical formula of the solid. In many cases the molecule of a metal in dilute solution in another metal is either monatomic, or forms a compound molecule with the solvent containing one atom of the dissolved metal, in which case the molecular depression is given by putting the atomic weight for M. In other cases, as Cu, Hg, Zn, in solution in cadmium, the depression of the F.P. per atom, according to Heycock and Neville, is only half as great, which would imply a diatomic molecule. Similarly As and Au in Cd appear to be triatomic, and Sn in Pb tetratomic. Intermediate cases may occur in which different molecules exist together in equilibrium in proportions which vary according to the temperature and concentration. The most familiar case is that of an electrolyte, in which the molecule of the dissolved substance is partly dissociated into ions. In such cases the degree of dissociation may be estimated by observing the depression of the F.P., but the results obtained cannot always be reconciled with those deduced by other methods, such as measurement of electrical conductivity, and there are many difficulties which await satisfactory interpretation.
Exactly similar relations to (8) and (9) apply to changes of boiling point or vapour pressure produced by substances in solution (see Vaporization), the laws of which are very closely connected with the corresponding phenomena of fusion; but the consideration of the vapour phase may generally be omitted in dealing with the fusion of mixtures where the vapour pressure of either constituent is small.
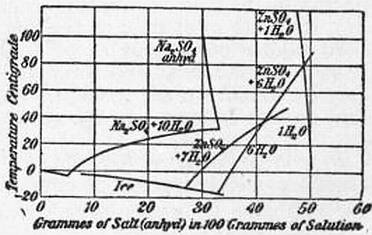 |
| Fig. 3.—Solubility Curves of Hydrates. |
8. Hydrates.—The simple case of a freezing point curve, illustrated in fig. 1, is generally modified by the occurrence of compounds of a character analogous to hydrates of soluble salts, in which the dissolved substance combines with one or more molecules of the solvent. These hydrates may exist as compound molecules in the solution, but their composition cannot be demonstrated unless they can be separated in the solid state. Corresponding to each crystalline hydrate there is generally a separate branch of the solubility curve along which the crystals of the hydrate are in equilibrium with the saturated solution. At any given temperature the hydrate possessing the least solubility is the most stable. If two are present in contact with the same solution, the more soluble will dissolve, and the less soluble will be formed at its expense until the conversion is complete. The two hydrates cannot be in equilibrium with the same solution except at the temperature at which their solubilities are equal, i.e. at the point where the corresponding curves of solubility intersect. This temperature is called the “Transition Point.” In the case of ZnSO4, as shown in fig. 3, the heptahydrate, with seven molecules of water, is the least soluble hydrate at ordinary temperatures, and is generally deposited from saturated solutions. Above 39° C., however, the hexahydrate, with six molecules, is less soluble, and a rapid conversion of the hepta- into the hexahydrate occurs if the former is heated above the transition point. The solubility of the hexahydrate is greater than that of the heptahydrate below 39°, but increases more slowly with rise of temperature. At about 80° C. the hexahydrate gives place to the monohydrate, which dissolves in water with evolution of heat, and diminishes in solubility with rise of temperature. Intermediate hydrates exist, but they are more soluble, and cannot be readily isolated. Both the mono- and hexahydrates are capable of existing in equilibrium with saturated solutions at temperatures far below their transition points, provided that the less soluble hydrate is not present in the crystalline form. The solubility curves can therefore be traced, as in fig. 3, over an extended range of temperature. The equilibrium of each hydrate with the solvent, considered separately, would present a diagram of two branches similar to fig. 1, but as a rule only a small portion of each curve can be realized, and the complete solubility curve, as experimentally determined, is composed of a number of separate pieces corresponding to the ranges of minimum solubility of different hydrates. Failure to recognize this, coupled with the fact that in strong and viscous solutions the state of equilibrium is but slowly attained, is the probable explanation of the remarkable discrepancies existing in many recorded data of solubility.
Transition Points of Hydrates.
| Na2CrO4·10H2O | 19.9° | NaBr·2H20 | 50.7° |
| Na2SO4·10H2O | 32.4° | MnCl2·4H2O | 57.8° |
| Na2CO3·10H2O | 35.1° | Na3PO4·12H2O | 73.4° |
| Na2S2O3·5H2O | 48.0° | Ba(OH)2·8H2O | 77.9° |
The transition points of the hydrates given in the above list (Richards, Proc. Amer. Acad., 1899, 34, p. 277) afford well-marked constant temperatures which can be utilized as fixed points for experimental purposes.
9. Formation of Mixed Crystals.—An important exception to the general type already described, in which the addition of a dissolved substance lowers the F.P. of the solvent, is presented by the formation of mixed crystals, or “solid solutions,” in which the solvent and solute occur mixed in varying proportions. This isomorphous replacement of one substance by another, in the same crystal with little or no change of form, has long been known and studied in the case of minerals and salts, but the relations between composition and melting-point have seldom been investigated, and much still remains obscure. In this case the process of freezing does not necessitate the performance of work of separation of the constituents of the solution, the F.P. is not necessarily depressed, and the effect cannot be calculated by the usual formula for dilute solutions. One of the simplest types of F.P. curve which may result from the occurrence of mixed crystals is illustrated by the case of alloys of gold and silver, or gold and platinum, in which the F.P. curve is nearly a straight line joining the freezing-points of the constituents. The equilibrium between the solid and liquid, in both of which the two metals are capable of mixing in all proportions, bears in this case an obvious and close analogy to the equilibrium between a mixed liquid (e.g. alcohol and water) and its vapour. In the latter case, as is well known, the vapour will contain a larger proportion of the more volatile constituent. Similarly in the case of the formation of mixed crystals, the liquid should contain a larger proportion of the more fusible constituent than the solid with which it is in equilibrium. The composition of the crystals which are being deposited at any moment will, therefore, necessarily change as solidification proceeds, following the change in the composition of the liquid, and the temperature will fall until the last portions of the liquid to solidify will consist chiefly of the more fusible constituent, at the F.P. of which the solidification will be complete. If, however, as seems to be frequently the case, the composition of the solid and liquid phases do not greatly differ from each other, the greater part of the solidification will occur within a comparatively small range of temperature, and the initial F.P. of the alloy will be well marked. It is possible in this case to draw a second curve representing the composition of the solid phase which is in equilibrium with the liquid at any temperature. This curve will not represent the average composition of the crystals, but that of the outer coating only which is in equilibrium with the liquid at the moment. H.W.B. Roozeboom (Zeit. Phys. Chem. xxx. p. 385) has attempted to classify some of the possible cases which may occur in the formation of mixed crystals on the basis of J.W. Gibbs’s thermodynamic potential, the general properties of which may be qualitatively deduced from a consideration of observed phenomena. But although this method may enable us to classify different types, and even to predict results in a qualitative manner, it does not admit of numerical calculation similar to equation (8), as the Gibbs’s function itself is of a purely abstract nature and its form is unknown. There is no doubt that the formation of mixed crystals may explain many apparent anomalies in the study of F.P. curves. The whole subject has been most fruitful of results in recent years, and appears full of promise for the future.
For further details in this particular branch the reader may consult a report by Neville (Brit. Assoc. Rep., 1900), which contains numerous references to original papers by Roberts-Austen, Le Chatelier, Roozeboom and others. For the properties of solutions see Solution.