

Diffusion (from the Lat. diffundere; dis-, asunder, and fundere, to pour out), in general, a spreading out, scattering or circulation; in physics the term is applied to a special phenomenon, treated below.
1. General Description.—When two different substances are placed in contact with each other they sometimes remain separate, but in many cases a gradual mixing takes place. In the case where both the substances are gases the process of mixing continues until the result is a uniform mixture. In other cases the proportions in which two different substances can mix lie between certain fixed limits, but the mixture is distinguished from a chemical compound by the fact that between these limits the composition of the mixture is capable of continuous variation, while in chemical compounds, the proportions of the different constituents can only have a discrete series of numerical values, each different ratio representing a different compound. If we take, for example, air and water in the presence of each other, air will become dissolved in the water, and water will evaporate into the air, and the proportions of either constituent absorbed by the other will vary continuously. But a limit will come when the air will absorb no more water, and the water will absorb no more air, and throughout the change a definite surface of separation will exist between the liquid and the gaseous parts. When no surface of separation ever exists between two substances they must necessarily be capable of mixing in all proportions. If they are not capable of mixing in all proportions a discontinuous change must occur somewhere between the regions where the substances are still unmixed, thus giving rise to a surface of separation.
The phenomena of mixing thus involves the following processes:—(1) A motion of the substances relative to one another throughout a definite region of space in which mixing is taking place. This relative motion is called “diffusion.” (2) The passage of portions of the mixing substances across the surface of separation when such a surface exists. These surface actions are described under various terms such as solution, evaporation, condensation and so forth. For example, when a soluble salt is placed in a liquid, the process which occurs at the surface of the salt is called “solution,” but the salt which enters the liquid by solution is transported from the surface into the interior of the liquid by “diffusion.”
Diffusion may take place in solids, that is, in regions occupied by matter which continues to exhibit the properties of the solid state. Thus if two liquids which can mix are separated by a membrane or partition, the mixing may take place through the membrane. If a solution of salt is separated from pure water by a sheet of parchment, part of the salt will pass through the parchment into the water. If water and glycerin are separated in this way most of the water will pass into the glycerin and a little glycerin will pass through in the opposite direction, a property frequently used by microscopists for the purpose of gradually transferring minute algae from water into glycerin. A still more interesting series of examples is afforded by the passage of gases through partitions of metal, notably the passage of hydrogen through platinum and palladium at high temperatures. When the process is considered with reference to a membrane or partition taken as a whole, the passage of a substance from one side to the other is commonly known as “osmosis” or “transpiration” (see Solution), but what occurs in the material of the membrane itself is correctly described as diffusion.
Simple cases of diffusion are easily observed qualitatively. If a solution of a coloured salt is carefully introduced by a funnel into the bottom of a jar containing water, the two portions will at first be fairly well defined, but if the mixture can exist in all proportions, the surface of separation will gradually disappear; and the rise of the colour into the upper part and its gradual weakening in the lower part, may be watched for days, weeks or even longer intervals. The diffusion of a strong aniline colouring matter into the interior of gelatine is easily observed, and is commonly seen in copying apparatus. Diffusion of gases may be shown to exist by taking glass jars containing vapours of hydrochloric acid and ammonia, and placing them in communication with the heavier gas downmost. The precipitation of ammonium chloride shows that diffusion exists, though the chemical action prevents this example from forming a typical case of diffusion. Again, when a film of Canada balsam is enclosed between glass plates, the disappearance during a few weeks of small air bubbles enclosed in the balsam can be watched under the microscope.
In fluid media, whether liquids or gases, the process of mixing is greatly accelerated by stirring or agitating the fluids, and liquids which might take years to mix if left to themselves can thus be mixed in a few seconds. It is necessary to carefully distinguish the effects of agitation from those of diffusion proper. By shaking up two liquids which do not mix we split them up into a large number of different portions, and so greatly increase the area of the surface of separation, besides decreasing the thicknesses of the various portions. But even when we produce the appearance of a uniform turbid mixture, the small portions remain quite distinct. If however the fluids can really mix, the final process must in every case depend on diffusion, and all we do by shaking is to increase the sectional area, and decrease the thickness of the diffusing portions, thus rendering the completion of the operation more rapid. If a gas is shaken up in a liquid the process of absorption of the bubbles is also accelerated by capillary action, as occurs in an ordinary sparklet bottle. To state the matter precisely, however finely two fluids have been subdivided by agitation, the molecular constitution of the different portions remains unchanged. The ultimate process by which the individual molecules of two different substances become mixed, producing finally a homogeneous mixture, is in every case diffusion. In other words, diffusion is that relative motion of the molecules of two different substances by which the proportions of the molecules in any region containing a finite number of molecules are changed.
In order, therefore, to make accurate observations of diffusion in fluids it is necessary to guard against any cause which may set up currents; and in some cases this is exceedingly difficult. Thus, if gas is absorbed at the upper surface of a liquid, and if the gaseous solution is heavier than the pure liquid, currents may be set up, and a steady state of diffusion may cease to exist. This has been tested experimentally by C. G. von Hüfner and W. E. Adney. The same thing may happen when a gas is evolved into a liquid at the surface of a solid even if no bubbles are formed; thus if pieces of aluminium are placed in caustic soda, the currents set up by the evolution of hydrogen are sufficient to set the aluminium pieces in motion, and it is probable that the motions of the Diatomaceae are similarly caused by the evolution of oxygen. In some pairs of substances diffusion may take place more rapidly than in others. Of course the progress of events in any experiment necessarily depends on various causes, such as the size of the containing vessels, but it is easy to see that when experiments with different substances are carried out under similar conditions, however these “similar conditions” be defined, the rates of diffusion must be capable of numerical comparison, and the results must be expressible in terms of at least one physical quantity, which for any two substances can be called their coefficient of diffusion. How to select this quantity we shall see later.
2 Quantitative Methods of observing Diffusion.—The simplest plan of determining the progress of diffusion between two liquids would be to draw off and examine portions from different strata at some stage in the process; the disturbance produced would, however, interfere with the subsequent process of diffusion, and the observations could not be continued. By placing in the liquid column hollow glass beads of different average densities, and observing at what height they remain suspended, it is possible to trace the variations of density of the liquid column at different depths, and different times. In this method, which was originally introduced by Lord Kelvin, difficulties were caused by the adherence of small air bubbles to the beads.
In general, optical methods are the most capable of giving exact results, and the following may be distinguished, (a) By refraction in a horizontal plane. If the containing vessel is in the form of a prism, the deviation of a horizontal ray of light in passing through the prism determines the index of refraction, and consequently the density of the stratum through which the ray passes, (b) By refraction in a vertical plane. Owing to the density varying with the depth, a horizontal ray entering the liquid also undergoes a small vertical deviation, being bent downwards towards the layers of greater density. The observation of this vertical deviation determines not the actual density, but its rate of variation with the depth, i.e. the “density gradient” at any point, (c) By the saccharimeter. In the cases of solutions of sugar, which cause rotation of the plane of polarized light, the density of the sugar at any depth may be determined by observing the corresponding angle of rotation, this was done originally by W. Voigt.
3. Elementary Definitions of Coefficient of Diffusion.—The simplest case of diffusion is that of a substance, say a gas, diffusing in the interior of a homogeneous solid medium, which remains at rest, when no external forces act on the system. We may regard it as the result of experience that: (1) if the density of the diffusing substance is everywhere the same no diffusion takes place, and (2) if the density of the diffusing substance is different at different points, diffusion will take place from places of greater to those of lesser density, and will not cease until the density is everywhere the same. It follows that the rate of flow of the diffusing substance at any point in any direction must depend on the density gradient at that point in that direction, i.e. on the rate at which the density of the diffusing substance decreases as we move in that direction. We may define the coefficient of diffusion as the ratio of the total mass per unit area which flows across any small section, to the rate of decrease of the density per unit distance in a direction perpendicular to that section.
In the case of steady diffusion parallel to the axis of x, if ρ be the density of the diffusing substance, and q the mass which flows across a unit of area in a plane perpendicular to the axis of x, then the density gradient is -dρ/dx and the ratio of q to this is called the “coefficient of diffusion.” By what has been said this ratio remains finite, however small the actual gradient and flow may be., and it is natural to assume, at any rate as a first approximation, that it is constant as far as the quantities in question are concerned. Thus if the coefficient of diffusion be denoted by K we have q= -K(dρ/dx).
Further, the rate at which the quantity of substance is increasing in an element between the distances x and x+dx is equal to the difference of the rates of flow in and out of the two faces, whence as in hydrodynamics, we have dρ/dt =-dq/dx.
It follows that the equation of diffusion in this case assumes the form
| dρ | = | d | ( K | dρ | ), |
| dt | dx | dx |
which is identical with the equations representing conduction of heat, flow of electricity and other physical phenomena. For motion in three dimensions we have in like manner
| dρ | = | d | ( K | dρ | ) + | d | ( K | dρ | ) + | d | ( K | dρ | ); |
| dt | dx | dx | dy | dy | dz | dz |
and the corresponding equations in electricity and heat for anisotropic substances would be available to account for any parallel phenomena, which may arise, or might be conceived, to exist in connexion with diffusion through a crystalline solid.
In the case of a very dilute solution, the coefficient of diffusion of the dissolved substance can be defined in the same way as when the diffusion takes place in a solid, because the effects of diffusion will not have any perceptible influence on the solvent, and the latter may therefore be regarded as remaining practically at rest. But in most cases of diffusion between two fluids, both of the fluids are in motion, and hence there is far greater difficulty in determining the motion, and even in defining the coefficient of diffusion. It is important to notice in the first instance, that it is only the relative motion of the two substances which constitutes diffusion. Thus when a current of air is blowing, under ordinary circumstances the changes which take place are purely mechanical, and do not depend on the separate diffusions of the oxygen and nitrogen of which the air is mainly composed. It is only when two gases are flowing with unequal velocity, that is, when they have a relative motion, that these changes of relative distribution, which are called diffusion, take place. The best way out of the difficulty is to investigate the separate motions of the two fluids, taking account of the mechanical actions exerted on them, and supposing that the mutual action of the fluids causes either fluid to resist the relative motion of the other.
4. The Coefficient of Resistance.—Let us call the two diffusing fluids A and B. If B were absent, the motion of the fluid A would be determined entirely by the variations of pressure of the fluid A, and by the external forces, such as that due to gravity acting on A. Similarly if A were absent, the motion of B would be determined entirely by the variations of pressure due to the fluid B, and by the external forces acting on B. When both fluids are mixed together, each fluid tends to resist the relative motion of the other, and by the law of equality of action and reaction, the resistance which A experiences from B is everywhere equal and opposite to the resistance which B experiences from A. If the amount of this resistance per unit volume be divided by the relative velocity of the two fluids, and also by the product of their densities, the quotient is called the “coefficient of resistance.” If then ρ1, ρ2 are the densities cf the two fluids, u1, u2 their velocities, C the coefficient of resistance, then the portion of the fluid A contained in a small element of volume v will experience from the fluid B a resistance Cρ1ρ2v(u1 − u2), and the fluid B contained in the same volume element will experience from the fluid A an equal and opposite resistance, Cρ1ρ2v(u2 − u1).
This definition implies the following laws of resistance to diffusion, which must be regarded as based on experience, and not as self-evident truths: (1) each fluid tends to assume, so far as diffusion is concerned, the same equüibrium distribution that it would assume if its motion were unresisted by the presence of the other fluid. (Of course, the mutual attraction of gravitation of the two fluids might affect the final distribution, but this is practically negligible. Leaving such actions as this out of account the following statement is correct.) In a state of equilibrium, the density of each fluid at any point thus depends only on the partial pressure of that fluid alone, and is the same as if the other fluids were absent. It does not depend on the partial pressures of the other fluids. If this were not the case, the resistance to diffusion would be analogous to friction, and would contain terms which were independent of the relative velocity u2 − u1. (2) For slow motions the resistance to diffusion is (approximately at any rate) proportional to the relative velocity. (3) The coefficient of resistance C is not necessarily always constant; it may, for example, and, in general, does, depend on the temperature.
If we form the equations of hydrodynamics for the different fluids occurring in any mixture, taking account of diffusion, but neglecting viscosity, and using suffixes 1, 2 to denote the separate fluids, these assume the form given by James Clerk Maxwell (“Diffusion,” in Ency. Brit., 9th ed.):—
| ρ1 | Du1 | + | dp1 | − X1ρ1 + C12ρ1ρ2(u1 − u2) + &c. = 0, |
| Dt | dx |
where
| Du1 | = | du1 | + u1 | du1 | + v1 | du1 | + w1 | du1 | , |
| Dt | dt | dx | dy | dz |
and these equations imply that when diffusion and other motions cease, the fluids satisfy the separate conditions of equilibrium dp1/dx − X1ρ1 = 0. The assumption made in the following account is that terms such as Du1/Dt may be neglected in the cases considered.
A further property based on experience is that the motions set up in a mixture by diffusion are very slow compared with those set up by mechanical actions, such as differences of pressure. Thus, if two gases at equal temperature and pressure be allowed to mix by diffusion, the heavier gas being below the lighter, the process will take a long time; on the other hand, if two gases, or parts of the same gas, at different pressures be connected, equalization of pressure will take place almost immediately. It follows from this property that the forces required to overcome the “inertia” of the fluids in the motions due to diffusion are quite imperceptible. At any stage of the process, therefore, any one of the diffusing fluids may be regarded as in equilibrium under the action of its own partial pressure, the external forces to which it is subjected and the resistance to diffusion of the other fluids.
5. Slow Diffusion of two Gases. Relation between the Coefficients of Resistance and of Diffusion.—We now suppose the diffusing substances to be two gases which obey Boyle’s law, and that diffusion takes place in a closed cylinder or tube of unit sectional area at constant temperature, the surfaces of equal density being perpendicular to the axis of the cylinder, so that the direction of diffusion is along the length of the cylinder, and we suppose no external forces, such as gravity, to act on the system.
The densities of the gases are denoted by ρ1, ρ2, their velocities of diffusion by u1, u2, and if their partial pressures are p1, p2, we have by Boyle’s law p1 = k1ρ1, p2 = k2ρ2, where k1,k2 are constants for the two gases, the temperature being constant. The axis of the cylinder is taken as the axis of x.
From the considerations of the preceding section, the effects of inertia of the diffusing gases may be neglected, and at any instant of the process either of the gases is to be treated as kept in equilibrium by its partial pressure and the resistance to diffusion produced by the other gas. Calling this resistance per unit volume R, and putting R = Cρ1ρ2(u1 − u2), where C is the coefficient of resistance, the equations of equilibrium give
| dp1 | + Cρ1ρ2(u1 − u2) = 0, and | dp2 | + Cρ1ρ2(u2 − u1) = 0 (1) |
| dx | dx |
These involve
| dp1 | + | dp2 | = 0 or p1 + p2 = P (2) |
| dx | dx |
where P is the total pressure of the mixture, and is everywhere constant, consistently with the conditions of mechanical equilibrium.
Now dp1/dx is the pressure-gradient of the first gas, and is, by Boyle’s law, equal to k1 times the corresponding density-gradient. Again ρ1u1 is the mass of gas flowing across any section per unit time, and k1ρ1u1 or p1u1 can be regarded as representing the flux of partial pressure produced by the motion of the gas. Since the total pressure is everywhere constant, and the ends of the cylinder are supposed fixed, the fluxes of partial pressure due to the two gases are equal and opposite, so that
p1u1 + p2u2 = 0 or k1ρ1u1 + k2ρ2u2 = 0 (3).
From (2) (3) we find by elementary algebra
u1/p2 = −u2/p1 = (u1 − u2)/(p1 + p2) = (u1 − u2)/P,
and therefore
p2u1 = −p2u2 = p1p2(u1 − u2)/P = k1k2ρ1ρ2(u1 − u2)/P
Hence equations (1) (2) gives
| dp1 | + | CP | (p1u1) = 0, and | dp2 | + | CP | (p2u2) = 0; |
| dx | k1k2 | dx | k1k2 |
whence also substituting p1 = k1ρ1, p2 = k2ρ2, and by transposing
| ρ1u1 = − | k1k2 | dρ1 | , and ρ2u2 = − | k1k2 | dρ2 | . |
| CP | dx | CP | dx |
We may now define the “coefficient of diffusion” of either gas as the ratio of the rate of flow of that gas to its density-gradient. With this definition, the coefficients of diffusion of both the gases in a mixture are equal, each being equal to k1k2/CP. The ratios of the fluxes of partial pressure to the corresponding pressure-gradients are also equal to the same coefficient. Calling this coefficient K, we also observe that the equations of continuity for the two gases are
| dρ1 | + | d(ρ1u1) | = 0, and | dρ2 | + | d(ρ2u2) | = 0, |
| dt | dx | dt | dx |
leading to the equations of diffusion
| dρ1 | = | d | (K | dρ1 | ), and | dρ2 | = | d | (K | dρ2 | ), |
| dt | dx | dx | dt | dx | dx |
exactly as in the case of diffusion through a solid.
If we attempt to treat diffusion in liquids by a similar method, it is, in the first place, necessary to define the “partial pressure” of the components occurring in a liquid mixture. This leads to the conception of “osmotic pressure,” which is dealt with in the article Solution. For dilute solutions at constant temperature, the assumption that the osmotic pressure is proportional to the density, leads to results agreeing fairly closely with experience, and this fact may be represented by the statement that a substance occurring in a dilute solution behaves like a perfect gas.
6. Relation of the Coefficient of Diffusion to the Units of Length and Time.—We may write the equation defining K in the form
| −K × | I | dρ | . |
| ρ | dx |
Here −dρ/ρdx represents the “percentage rate” at which the density decreases with the distance x; and we thus see that the coefficient of diffusion represents the ratio of the velocity of flow to the percentage rate at which the density decreases with the distance measured in the direction of flow. This percentage rate being of the nature of a number divided by a length, and the velocity being of the nature of a length divided by a time, we may state that K is of two dimensions in length and −1 in time, i.e. dimensions L²/T.
Example 1. Taking K = 0.1423 for carbon dioxide and air (at temperature 0° C. and pressure 76 cm. of mercury) referred to a centimetre and a second as units, we may interpret the result as follows:—Supposing in a mixture of carbon dioxide and air, the density of the carbon dioxide decreases by, say, 1, 2 or 3% of itself in a distance of 1 cm., then the corresponding velocities of the diffusing carbon dioxide will be respectively 0.01, 0.02 and 0.03 times 0.1423, that is, 0.001423, 0.002846 and 0.004269 cm. per second in the three cases.
Example 2. If we wished to take a foot and a second as our units, we should have to divide the value of the coefficient of diffusion in Example 1 by the square of the number of centimetres in 1 ft., that is, roughly speaking, by 900, giving the new value of K = 0.00016 roughly.
7. Numerical Values of the Coefficient of Diffusion.—The table on p. 258 gives the values of the coefficient of diffusion of several of the principal pairs of gases at a pressure of 76 cm. of mercury, and also of a number of other substances. In the gases the centimetre and second are taken as fundamental units, in other cases the centimetre and day.
8. Irreversible Changes accompanying Diffusion.—The diffusion of two gases at constant pressure and temperature is a good example of an “irreversible process.” The gases always tend to mix, never to separate. In order to separate the gases a change must be effected in the external conditions to which the mixture is subjected, either by liquefying one of the gases, or by separating them by diffusion through a membrane, or by bringing other outside influences to bear on them. In the case of liquids, electrolysis affords a means of separating the constituents of a mixture. Every such method involves some change taking place outside the mixture, and this change may be regarded as a “compensating transformation.” We thus have an instance of the property that every irreversible change leaves an indelible imprint somewhere or other on the progress of events in the universe. That the process of diffusion obeys the laws of irreversible thermodynamics (if these laws are properly stated) is proved by the fact that the compensating transformations required to separate mixed gases do not essentially involve anything but transformation of energy. The process of allowing gases to mix by diffusion, and then separating them by a compensating transformation, thus constitutes an irreversible cycle, the outside effects of which are that energy somewhere or other must be less capable of transformation than it was before the change. We express this fact by stating that an irreversible process essentially implies a loss of availability. To measure this loss we make use of the laws of thermodynamics, and in particular of Lord Kelvin’s statement that “It is impossible by means of inanimate material agency to derive mechanical effect from any portion of matter by cooling it below the temperature of the coldest of the surrounding objects.”
| Substances. | Temp. | K. | Author. |
| Carbon dioxide and air | 0°C. | 0.1423 cm²/sec. | J. Loschmidt. |
| ” ” hydrogen | 0°C. | 0.5558 ” | ” |
| ” ” oxygen | 0°C. | 0.1409 ” | ” |
| ” ” carbon monoxide | 0°C. | 0.1406 ” | ” |
| ” ” marsh gas (methane) | 0°C. | 0.1586 ” | ” |
| ” ” nitrous oxide | 0°C. | 0.0983 ” | ” |
| Hydrogen and oxygen | 0°C. | 0.7214 ” | ” |
| ” ” carbon monoxide | 0°C. | 0.6422 ” | ” |
| ” ” sulphur dioxide | 0°C. | 0.4800 ” | ” |
| Oxygen and carbon monoxide | 0°C. | 0.1802 ” | ” |
| Water and ammonia | 20°C. | 1.250 ” | G. Hüfner. |
| ” ” | 5°C. | 0.822 ” | ” |
| ” common salt (density 1.0269) | 0.355 ” | J. Graham. | |
| ” ” ” | 14.33°C. | 1.020, 0.996, 0.972, 0.932 cm²/day. | F. Heimbrodt. |
| ” zinc sulphate (0.312 gm/cm³) | 0.1162 cm²/day. | W. Seitz. | |
| ” zinc sulphate (normal) | 0.2355 ” | ” | |
| ” zinc acetate (double normal) | 0.1195 ” | ” | |
| ” zinc formate (half normal) | 0.4654 ” | ” | |
| ” cadmium sulphate (double normal) | · · | 0.2456 ” | ” |
| ” glycerin (1⁄8n, ½n, 7⁄8n, 1.5n) | 10.14°C. | 0.356, 0.350, 0.342, 0.315 cm²/day. | F. Heimbrodt. |
| ” urea ” ” | 14.83°C. | 0.973, 0.946, 0.926, 0.883 cm²/day. | ” |
| ” hydrochloric acid | 14.30°C. | 2.208, 2.331, 2.480 cm²/day. | ” |
| Gelatin 20% and ammonia | 17°C. | 127.1 cm²/day. | A. Hagenbach. |
| ” ” carbon dioxide | · · | 0.845 ” | ” |
| ” ” nitrous oxide | · · | 0.509 ” | ” |
| ” ” oxygen | · · | 0.230 ” | ” |
| ” ” hydrogen | · · | 0.0565 ” | ” |
Let us now assume that we have any syste m such as the gases above considered, and that it is in the presence of an indefinitely extended medium which we shall call the “auxiliary medium.” If heat be taken from any part of the system, only part of this heat can be converted into work by means of thermodynamic engines; and the rest will be given to the auxiliary medium, and will constitute unavailable energy or waste. To understand what this means, we may consider the case of a condensing steam engine. Only part of the energy liberated by the combustion of the coal is available for driving the engine, the rest takes the form of heat imparted to the condenser. The colder the condenser the more efficient is the engine, and the smaller is the quantity of waste.
The amount of unavailable energy associated with any given transformation is proportional to the absolute temperature of the auxiliary medium. When divided by that temperature the quotient is called the change of “entropy” associated with the given change (see Thermodynamics). Thus if a body at temperature T receives a quantity of heat Q, and if T0 is the temperature of the auxiliary medium, the quantity of work which could be obtained from Q by means of ideal thermodynamic engines would be Q(1 − T0/T), and the balance, which is QT0/T, would take the form of unavailable or waste energy given to the medium. The quotient of this, when divided by T0, is Q/T, and this represents the quantity of entropy associated with Q units of heat at temperature T.
Any irreversible change for which a compensating transformation of energy exists represents, therefore, an increase of unavailable energy, which is measurable in terms of entropy. The increase of entropy is independent of the temperature of the auxiliary medium. It thus affords a measure of the extent to which energy has run to waste during the change. Moreover, when a body is heated, the increase of entropy is the factor which determines how much of the energy imparted to the body is unavailable for conversion into work under given conditions. In all cases we have
| increase of unavailable energy | = increase of entropy. |
| temperature of auxiliary medium |
When diffusion takes place between two gases inside a closed vessel at uniform pressure and temperature no energy in the form of heat or work is received from without, and hence the entropy gained by the gases from without is zero. But the irreversible processes inside the vessel may involve a gain of entropy, and this can only be estimated by examining by what means mixed gases can be separated, and, in particular, under what conditions the process of mixing and separating the gases could (theoretically) be made reversible.
9. Evidence derived from Liquefaction of one or both of the Gases.—The gases in a mixture can often be separated by liquefying, or even solidifying, one or both of the components. In connexion with this property we have the important law according to which “The pressure of a vapour in equilibrium with its liquid depends only on the temperature and is independent of the pressures of any other gases or vapours which may be mixed with it.” Thus if two closed vessels be taken containing some water and one be exhausted, the other containing air, and if the temperatures be equal, evaporation will go on until the pressure of the vapour in the exhausted vessel is equal to its partial pressure in the other vessel, notwithstanding the fact that the total pressure in the latter vessel is greater by the pressure of the air.
To separate mixed gases by liquefaction, they must be compressed and cooled till one separates in the form of a liquid. If no changes are to take place outside the system, the separate components must be allowed to expand until the work of expansion is equal to the work of compression, and the heat given out in compression is reabsorbed in expansion. The process may be made as nearly reversible as we like by performing the operations so slowly that the substances are practically in a state of equilibrium at every stage. This is a consequence of an important axiom in thermodynamics according to which “any small change in the neighbourhood of a state of equilibrium is to a first approximation reversible.”
Suppose now that at any stage of the compression the partial pressures of the two gases are p1 and p2, and that the volume is changed from V to V − dV. The work of compression is (p1 + p2)dV, and this work will be restored at the corresponding stage if each of the separated gases increases in volume from V − dV to V. The ultimate state of the separated gases will thus be one in which each gas occupies the volume V originally occupied by the mixture.
We may now obtain an estimate of the amount of energy rendered unavailable by diffusion. We suppose two gases occupying volumes V1 and V2 at equal pressure p to mix by diffusion, so that the final volume is V1 + V2. Then if before mixing each gas had been allowed to expand till its volume was V1 + V2, work would have been done in the expansion, and the gases could still have been mixed by a reversal of the process above described. In the actual diffusion this work of expansion is lost, and represents energy rendered unavailable at the temperature at which diffusion takes place. When divided by that temperature the quotient gives the increase of entropy. Thus the irreversible processes, and, in particular, the entropy changes associated with diffusion of two gases at uniform pressure, are the same as would take place if each of the gases in turn were to expand by rushing into a vacuum, till it occupied the whole volume of the mixture. A more rigorous proof involves considerations of the thermodynamic potentials, following the methods of J. Willard Gibbs (see Energetics).
Another way in which two or more mixed gases can be separated is by placing them in the presence of a liquid which can freely absorb one of the gases, but in which the other gas or gases are insoluble. Here again it is found by experience that when equilibrium exists at a given temperature between the dissolved and undissolved portions of the first gas, the partial pressure of that gas in the mixture depends on the temperature alone, and is independent of the partial pressures of the insoluble gases with which it is mixed, so that the conclusions are the same as before.
10. Diffusion through a Membrane or Partition. Theory of the semi-permeable Membrane.—It has been pointed out that diffusion of gases frequently takes place in the interior of solids; moreover, different gases behave differently with respect to the same solid at the same temperature. A membrane or partition formed of such a solid can therefore be used to effect a more or less complete separation of gases from a mixture. This method is employed commercially for extracting oxygen from the atmosphere, in particular for use in projection lanterns where a high degree of purity is not required. A similar method is often applied to liquids and solutions and is known as “dialysis.”
In such cases as can be tested experimentally it has been found that a gas always tends to pass through a membrane from the side where its density, and therefore its partial pressure, is greater to the side where it is less; so that for equilibrium the partial pressures on the two sides must be equal. This result is unaffected by the presence of other gases on one or both sides of the membrane. For example, if different gases at the same pressure are separated by a partition through which one gas can pass more rapidly than the other, the diffusion will give rise to a difference of pressure on the two sides, which is capable of doing mechanical work in moving the partition. In evidence of this conclusion Max Planck quotes a test experiment made by him in the Physical Institute of the university of Munich in 1883, depending on the fact that platinum foil at white heat is permeable to hydrogen but impermeable to air, so that if a platinum tube filled with hydrogen be heated the hydrogen will diffuse out, leaving a vacuum.
The details of the experiment may be quoted here:—“A glass tube of about 5 mm. internal diameter, blown out to a bulb at the middle, was provided with a stop-cock at one end. To the other a platinum tube 10 cm. long was fastened, and closed at the end. The whole tube was exhausted by a mercury pump, filled with hydrogen at ordinary atmospheric pressure, and then closed. The closed end of the platinum portion was then heated in a horizontal position by a Bunsen burner. The connexion between the glass and platinum tubes, having been made by means of sealing-wax, had to be kept cool by a continuous current of water to prevent the softening of the wax. After four hours the tube was taken from the flame, cooled to the temperature of the room, and the stop-cock opened under mercury. The mercury rose rapidly, almost completely filling the tube, proving that the tube had been very nearly exhausted.”
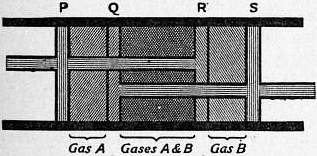 |
In order that diffusion through a membrane may be reversible so far as a particular gas is concerned, the process must take place so slowly that equilibrium is set up at every stage (see § 9 above). In order to separate one gas from another consistently with this condition it is necessary that no diffusion of the latter gas should accompany the process. The name “semi-permeable” is applied to an ideal membrane or partition through which one gas can pass, and which offers an insuperable barrier to any diffusion whatever of a second gas. By means of two semi-permeable partitions acting oppositely with respect to two different gases A and B these gases could be mixed or separated by reversible methods. The annexed figure shows a diagrammatic representation of the process.
We suppose the gases contained in a cylindrical tube; P, Q, R, S are four pistons, of which P and R are joined to one connecting rod, Q and S to another. P, S are impermeable to both gases; Q is semi-permeable, allowing the gas A to pass through but not B, similarly R allows the gas B to pass through but not A. The distance PR is equal to the distance QS, so that if the rods are pushed towards each other as far as they will go, P and Q will be in contact, as also R and S. Imagine the space RQ filled with a mixture of the two gases under these conditions. Then by slowly drawing the connecting rods apart until R, Q touch, the gas A will pass into the space PQ, and B will pass into the space RS, and the gases will finally be completely separated; similarly, by pushing the connecting rods together, the two gases will be remixed in the space RQ. By performing the operations slowly enough we may make the processes as nearly reversible as we please, so that no available energy is lost in either change. The gas A being at every instant in equilibrium on the two sides of the piston Q, its density, and therefore its partial pressure, is the same on both sides, and the same is true regarding the gas B on the two sides of R. Also no work is done in moving the pistons, for the partial pressures of B on the two sides of R balance each other, consequently, the resultant thrust on R is due to the gas A alone, and is equal and opposite to its resultant thrust on P, so that the connecting rods are at every instant in a state of mechanical equilibrium so far as the pressures of the gases A and B are concerned. We conclude that in the reversible separation of the gases by this method at constant temperature without the production or absorption of mechanical work, the densities and the partial pressures of the two separated gases are the same as they were in the mixture. These conclusions are in entire agreement with those of the preceding section. If this agreement did not exist it would be possible, theoretically, to obtain perpetual motion from the gases in a way that would be inconsistent with the second law of thermodynamics.
Most physicists admit, as Planck does, that it is impossible to obtain an ideal semi-permeable substance; indeed such a substance would necessarily have to possess an infinitely great resistance to diffusion for such gases as could not penetrate it. But in an experiment performed under actual conditions the losses of available energy arising from this cause would be attributable to the imperfect efficiency of the partitions and not to the gases themselves; moreover, these losses are, in every case, found to be completely in accordance with the laws of irreversible thermodynamics. The reasoning in this article being somewhat condensed the reader must necessarily be referred to treatises on thermodynamics for further information on points of detail connected with the argument. Even when he consults these treatises he may find some points omitted which have been examined in full detail at some time or other, but are not sufficiently often raised to require mention in print.
II. Kinetic Models of Diffusion.—Imagine in the first instance that a very large number of red balls are distributed over one half of a billiard table, and an equal number of white balls over the other half. If the balls are set in motion with different velocities in various directions, diffusion will take place, the red balls finding their way among the white ones, and vice versa; and the process will be retarded by collisions between the balls. The simplest model of a perfect gas studied in the kinetic theory of gases (see Molecule) differs from the above illustration in that the bodies representing the molecules move in space instead of in a plane, and, unlike billiard balls, their motion is unresisted, and they are perfectly elastic, so that no kinetic energy is lost either during their free motions, or at a collision.
The mathematical analysis connected with the application of the kinetic theory to diffusion is very long and cumbersome. We shall therefore confine our attention to regarding a medium formed of elastic spheres as a mechanical model, by which the most important features of diffusion can be illustrated. We shall assume the results of the kinetic theory, according to which:—(1) In a dynamical model of a perfect gas the mean kinetic energy of translation of the molecules represents the absolute temperature of the gas. (2) The pressure at any point is proportional to the product of the number of molecules in unit volume about that point into the mean square of the velocity. (The mean square of the velocity is different from but proportional to the square of the mean velocity, and in the subsequent arguments either of these two quantities can generally be taken.) (3) In a gas mixture represented by a mixture of molecules of unequal masses, the mean kinetic energies of the different kinds are equal.
Consider now the problem of diffusion in a region containing two kinds of molecules A and B of unequal mass. The molecules of A in the neighbourhood of any point will, by their motion, spread out in every direction until they come into collision with other molecules of either kind, and this spreading out from every point of the medium will give rise to diffusion. If we imagine the velocities of the A molecules to be equally distributed in all directions, as they would be in a homogeneous mixture, it is obvious that the process of diffusion will be greater, ceteris paribus, the greater the velocity of the molecules, and the greater the length of the free path before a collision takes place. If we assume consistently with this, that the coefficient of diffusion of the gas A is proportional to the mean value of W{a}l{a}, where w{a} is the velocity and l{a} is the length of the path of a molecule of A, this expression for the coefficient of diffusion is of the right dimensions in length and time. If, moreover, we observe that when diffusion takes place in a fixed direction, say that of the axis of x, it depends only on the resolved part of the velocity and length of path in that direction: this hypothesis readily leads to our taking the mean value of 1⁄3wala as the coefficient of diffusion for the gas A. This value was obtained by O. E. Meyer and others.
Unfortunately, however, it makes the coefficients of diffusion unequal for the two gases, a result inconsistent with that obtained above from considerations of the coefficient of resistance, and leading to the consequence that differences of pressure would be set up in different parts of the gas. To equalize these differences of pressure, Meyer assumed that a counter current is set up, this current being, of course, very slow in practice; and J. Stefan assumed that the diffusion of one gas was not affected by collisions between molecules of the same gas. When the molecules are mixed in equal proportions both hypotheses lead to the value 1⁄6([wala] + [wblb]), (square brackets denoting mean values). When one gas preponderates largely over the other, the phenomena of diffusion are too difficult of observation to allow of accurate experimental tests being made. Moreover, in this case no difference exists unless the molecules are different in size or mass.
Instead of supposing a velocity of translation added after the mathematical calculations have been performed, a better plan is to assume from the outset that the molecules of the two gases have small velocities of translation in opposite directions, superposed on the distribution of velocity, which would occur in a medium representing a gas at rest. When a collision occurs between molecules of different gases a transference of momentum takes place between them, and the quantity of momentum so transferred in one second in a unit of volume gives a dynamical measure of the resistance to diffusion. It is to be observed that, however small the relative velocity of the gases A and B, it plays an all-important part in determining the coefficient of resistance; for without such relative motion, and with the velocities evenly distributed in all directions, no transference of momentum could take place. The coefficient of resistance being found, the motion of each of the two gases may be discussed separately.
One of the most important consequences of the kinetic theory is that if the volume be kept constant the coefficient of diffusion varies as the square root of the absolute temperature. To prove this, we merely have to imagine the velocity of each molecule to be suddenly increased n fold; the subsequent processes, including diffusion, will then go on n times as fast; and the temperature T, being proportional to the kinetic energy, and therefore to the square of the velocity, will be increased n² fold. Thus K, the coefficient of diffusion, varies as √T.
The relation of K to the density when the temperature remains constant is more difficult to discuss, but it may be sufficient to notice that if the number of molecules is increased n fold, the chances of a collision are n times as great, and the distance traversed between collisions is (not therefore but as the result of more detailed reasoning) on the average 1/n of what it was before. Thus the free path, and therefore the coefficient of diffusion, varies inversely as the density, or directly as the volume. If the pressure p and temperature T be taken as variables, K varies inversely as p and directly as √T³.
Now according to the experiments first made by J. C. Maxwell and J. Loschmidt, it appeared that with constant density K was proportional to T more nearly than to √T. The inference is that in this respect a medium formed of colliding spheres fails to give a correct mechanical model of gases. It has been found by L. Boltzmann, Maxwell and others that a system of particles whose mutual actions vary according to the inverse fifth power of the distance between them represents more correctly the relation between the coefficient of diffusion and temperature in actual gases. Other recent theories of diffusion have been advanced by M. Thiesen, P. Langevin and W. Sutherland. On the other hand, J. Thovert finds experimental evidence that the coefficient of diffusion is proportional to molecular velocity in the cases examined of non-electrolytes dissolved in water at 18° at 2.5 grams per litre.
Bibliography.—The best introduction to the study of theories of diffusion is afforded by O. E. Meyer’s Kinetic Theory of Gases, translated by Robert E. Baynes (London, 1899). The mathematical portion, though sufficient for ordinary purposes, is mostly of the simplest possible character. Another useful treatise is R. Ruhlmann’s Handbuch der mechanischen Wärmetheorie (Brunswick, 1885). For a shorter sketch the reader may refer to J. C. Maxwell’s Theory of Heat, chaps, xix. and xxii., or numerous other treatises on physics. The theory of the semi-permeable membrane is discussed by M. Planck in his Treatise on Thermodynamics, English translation by A. Ogg (1903), also in treatises on thermodynamics by W. Voigt and other writers. For a more detailed study of diffusion in general the following papers may be consulted:—L. Boltzmann, “Zur Integration der Diffusionsgleichung,” Sitzung. der k. bayer. Akad math.-phys. Klasse (May 1894); T. des Coudres, “Diffusionsvorgänge in einem Zylinder,” Wied. Ann. lv. (1895), p. 213; J. Loschmidt, “Experimentaluntersuchungen über Diffusion,” Wien. Sitz. lxi., lxii. (1870); J. Stefan, “Gleichgewicht und ... Diffusion von Gasmengen,” Wien. Sitz. lxiii., “Dynamische Theorie der Diffusion,” Wien. Sitz. lxv. (April 1872); M. Toepler, “Gas-diffusion,” Wied. Ann. lviii. (1896), p. 599; A. Wretschko, “Experimentaluntersuchungen über die Diffusion von Gasmengen,” Wien. Sitz. lxii. The mathematical theory of diffusion, according to the kinetic theory of gases, has been treated by a number of different methods, and for the study of these the reader may consult L. Boltzmann, Vorlesungen über Gastheorie (Leipzig, 1896-1898); S. H. Burbury, Kinetic Theory of Gases (Cambridge, 1899), and papers by L. Boltzmann in Wien. Sitz. lxxxvi. (1882), lxxxvii. (1883); P. G. Tait, “Foundations of the Kinetic Theory of Gases,” Trans. R.S.E. xxxiii., xxxv., xxvi., or Scientific Papers, ii. (Cambridge, 1900). For recent work reference should be made to the current issues of Science Abstracts (London), and entries under the heading “Diffusion” will be found in the general index at the end of each volume.